Improving compliance in Indian Pharma industry
Improving compliance in Indian Pharma industry
- Ganadhish Kamat
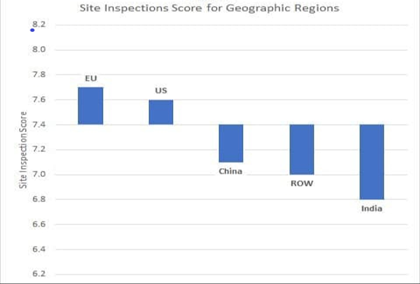
USFDA scores each site inspected by them based on the inspection outcome. The site inspection score provides one measure of a site’s compliance to CGMP regulations. Report on the State of Pharmaceutical Quality for 2019 published by CDER, included a summary of site inspection scores by different regions of the world. This summary showed that the average inspection score across the globe was 7.4. While the average scores of the sites in EU (7.7) and US (7.6) were above the global average, the average site scores in China (7.1), ROW (7.0) and India (6.8) were less, with Indian average being the least.
Indian sites also had large share of warning letters issued globally in last few years. The analysis of the warning letters issued to Indian sites since 2015 is given below –

After going through individual warning letters it is clear that the concerns raised in the warning letters were largely related to 5 areas mentioned below -
1. Data Reliability – This was a key issue cited in the WLs of 2015-16. There has been good improvement over a period of time, especially in the QC labs, but vulnerability still exists in other functions like manufacturing, engineering etc.
2. Inadequate Investigations – This continues to be a major issue in the industry. Either investigations are not thorough enough, or the root causes identified are not scientifically justifiable or supported by data. Repeat failures of the same type also indicate inadequacy of investigation and the CAPA.
3. Nitrosamines – In recent years, inadequate laboratory control has featured lot in the WLs. This was primarily on account of failure to establish appropriate specification for Nitrosamines because of lack of process understanding.
4. Inadequate Cleaning – This was cited in many recent WLs. This was not about cleaning validation but the failure to identify appropriate risks of contamination & cross-contamination and building appropriate controls in the cleaning procedures.
5. Sterility Assurance – Issues like inadequacies in media fills, aseptic practices, environment monitoring have been cited in the WLs of sterile facilities.
There is certainly need for a deep dive to understand reasons for such outcome and what Indian Pharma industry need to do to improve the situation.
Root cause analysis and suggestions for improvement–
The analysis given below is based on my personal experience of reviewing large number inspectional observations, Gemba walks, review of investigations, review of development reports, protocols, SOPs and other documents in various sites of many companies and interactions with large number of people.
1. Data reliability –
While most companies have complied to 21 CFR Part 11 / Annex 11 in the QC laboratories and implemented analytical data review (including electronic data and associated audit trails), still some loopholes exist in the systems. Unscrupulous elements make use of these loopholes and commit intentional violations. Some of the examples are –
1. Manual peak marking to adjust the results.
2. Unplugging the data cable to interrupt data transfer.
3. Repeat runs with slightly modified file name.
4. Storing of runs in different folders.
5. Changing of date and time.
6. Using other’s password.
Since a lot of different custom software are used in the QC lab, one solution does not fit all. Companies need to hire software experts who can thoroughly do the risk assessment to identify every possible loophole and put control measures. This assessment should be documented so that it can help in the future when any changes in the set up are planned. Periodic trending of all incidences or event logs should be done to identify emerging trends and any suspicious/repeat events should be thoroughly investigated.
The issue of part 11 non-compliance is common in R&D. Many times, it is wrongly believed that R&D is not in the purview of the audits and hence need not comply with GMPs. This is incorrect. Lot of data related to product development, which is important for assuring quality of the product, is generated in R&D and hence it is important to ensure reliability of such data even if the R&D labs are not audited. Some of the data generated is also included in the submissions and hence R&D can be subjected to audits. While all GMPs may not be followed in the R&D set up, data reliability must be assured in the R&D too and hence all R&D instruments also need to comply with Part 11. Apart from Part 11 compliance, controls such as documenting all the experiments, reasons for repeating experiments and not including certain data in submissions must be appropriately justified and documented.
Part 11 compliance has not been fully implemented in manufacturing due to various reasons. Many old equipment have PLC controls which do not comply with Part 11 requirements such as having individual login names and passwords, audit trails etc. The OEMs are also not supporting upgradation of these PLCs to meet current requirements. In such cases, it is important to do as risk assessment and establish additional controls like manual documentation of the activities carried out to maintain necessary paper audit trail. Wherever such equipment acquires process data, the data should be stored in non-editable format and should have all other security measures like back up, etc. It is better to eliminate any manual recording of such data and take direct printouts wherever possible to avoid duplication and entry errors. If for any reason the data is manually recorded, same should be periodically verified against the electronic data stored in the system to ensure that there is no mismatch.
Companies need to have policy of zero tolerance for data integrity violation and this should be constantly shared with the employees. Companies should join, to create data base of people indulging in unscrupulous activities, so that an employee terminated due to such reason from one company does not get employment in another place. This will work as a strong deterrent.
2. Inadequate investigations –
The main reasons for inadequate investigations in Pharma industry are given below –
• Lack of thorough product and process understanding.
QbD requires development of thorough product and process understanding by identifying all CMAs and CPPs, understanding their normal variability and the impact on the CQAs through DoE. This however is rarely done in the true sense. All these activities are completed to meet the filing requirement, but the data is not always scientifically evaluated to arrive at appropriate control strategy. This needs to undergo significant change. It is important to correctly document all the knowledge gathered in the drug development, including the failures if any in early experiments and transfer all the knowledge to the manufacturing sites. The knowledge gathering is a continuous process and as more batches are manufactured and more information is available, it is important to utilize that information to strengthen the control strategy.
• Lack of thorough understanding about manufacturing equipment.
The way product and process understanding is developed during design (development) stage, similarly it is important to develop complete understanding of equipment during its qualification. This should include understanding of –
- all the controls and set parameters of the equipment and their impact of the quality,
- design logic if the equipment is software controlled,
- alarms generated, their meaning, and actions to be taken for each type of alarm,
- environmental impact
- possible wear and tear and its impact on quality etc.
This knowledge of equipment and the product & process should be utilized to arrive at the final process design. This will result in fully establishing the process as required by 2011 validation guideline and eliminate the dependency on specific operators.
• Lack of availability of relevant information
Investigation often gets delayed due to lack of critical information which is required to reach the root cause. Information gathered for each process should be decided on the basis of process and machine understanding. Secondly, it is very important that the information available is reliable. To make the information reliable, the data of relevant parameters should be directly acquired from the equipment instead of relying on manual documentation. Apart from the process parameters, the following information is critical for investigations and should be captured in the batch records or machine records–
- Changes in machine setting (set parameters)
- Stoppages (even if minor)
- Alarms generated.
- Any abnormality observed etc.
• Reluctance to accept unfavorable data :-
Whenever an out of specification or out of trend result is observed, especially for established product, first thought which comes to mind is – ‘we have been making 1000s of batches without problem, this must be analytical error’. We must understand that any testing is based on small sample size and failure could be because of analytical error or could be due batch variability which might not have been detected earlier. The way we blindly accept the passing result, we also need to accept the failing result. Thorough and unbiased investigation is required to reach the root cause and take appropriate decision on the batch.
• Lack of systematic approach and no proper data analysis using statistical tools
Sometimes, due to pressure of completing the investigation quickly, people jump at the first cause where they see some correlation. Another approach which is commonly seen is performing trials based on some speculations and drawing conclusion about the root cause if trial results show positive outcomes. Such an approach may fail to identify the real root cause and the issue may resurface after some time. To get to the root cause of the problem, a systematic approach like DMAIC is required. Various statistical tools such as Brainstorming, 5 Whys, FMEA, Fault Tree Analysis, Fish-bone diagram, Regression analysis, Multivariate analysis, etc. can be used to channelize the thought process, minimize the data collection and experimentation and analysis of data to reach to meaningful conclusions. While data analysis tools can help in establishing correlation between the non-conformities and the parameters being evaluated, that alone is not enough to draw suitable conclusion. Scientific rationale needs to be understood for such correlation. To select appropriate tools for the investigation and for drawing right conclusions, investigators need to have thorough process understanding, machine understanding, understanding of the tools and analytical ability. In short, the best people in the company need to be entrusted with the investigations.
3. Nitrosamines –
Issue of Nitrosamines took the industry by surprise. It resulted in many recalls and several warning letters. This issue was triggered because of failure to identify all the potential risks in the process which could result in formation of Nitrosamines. This again points to the need for thorough process understanding and scientific risk assessments. While most pharmaceutical companies must have by now done the proper assessment of the processes to evaluate potential risk of Nitrosamines and established appropriate controls, this issue should not be restricted to Nitrosamines alone. Assessment needs to be extended to all GTIs in line with ICH M7. This assessment should be included in the API development process as one of the stage gate requirements. Similar assessment is also required for excipients.
4. Inadequate cleaning –
Some of warning letters under category of cleaning were related to inappropriate risk assessment of outsourced activity (solvent recovery) and failure to identify the potential of cross contamination with nitrosamines. With many companies outsourcing activities like solvent recovery, intermediate manufacturing, KSM manufacturing, etc., risk assessments for outsourced activities must be done in same depth as done in inhouse manufacturing sites.
Other warning letters related to cleaning were due to inability identify the risks of cross contamination. This points out to inadequate understanding of the equipment design to understand where contamination can remain and failure to design cleaning process to eliminate such possibility. Like manufacturing processes, cleaning processes need to be appropriately designed after thoroughly understanding equipment design and product nature. The process needs to be described in adequate detail to avoid operator to operator variability. Where possible, automation should be deployed for cleaning to achieve consistency. Manual processes cannot be validated in the true sense as there are far too many variables. In such cases, it is important to document each cleaning step, as done in case of manufacturing process, to avoid missing any step. Although equipment cleaning is very important activity to prevent cross-contamination, the design of processes is often left to the people with limited experience and are not thoroughly reviewed by experienced people. To make the cleaning process design robust, the process should be qualified by actual observation of entire process and matching each step with the process description. This activity should be done at adequately senior levels to make it effective.
Apart from poorly designed process, another critical reason for poor cleaning is inadequate supervision. Cleaning process is not given same level of importance as the manufacturing process and is often left to junior people with little to no supervision. Cleaning is often done during night shifts or on weekends when senior people are not present. This may result in some shortcuts if the activity is manual. Presence of supervisory personnel and managers should be distributed in all the shifts depending upon the level of activities.
Another reason for poor supervision is - most shopfloor managers remain busy in meetings and presentations and spend very little time on the shopfloor. Companies run multiple programs like efficiency improvement, cost improvement, compliance improvements etc. Each of these programs take significant bandwidth of shopfloor managers in gathering data, compiling information, preparing presentations for the review meetings, and attending the review meetings. While these initiatives are important, the way they are managed and reviewed need to change. Taking cue from Amazon and Apple, PowerPoint presentations should be stopped. Meetings should be kept short on the shopfloor, involving as less people as possible. The meetings should not be utilized to review the status of the project (that can be done through dashboards or pre-reads) but only to take decisions on specific issues which the group is not able to take. These changes are going to improve not just cleaning but all activities.
5. Sterility Assurance
As very little assurance about sterility can be provided by testing, the entire focus must go on process design and controls. Because of its criticality, design needs involvement of top experts in sterile product manufacturing, engineering, and microbiology. Every design aspect must be justified with sound scientific rationale and need to be well documented. These include but are not limited to -
1. Area design & classification
2. Cleaning and sanitization programs
3. Sterilization / depyrogenation processes
4. Gowning process
5. Process design
6. Hold times
7. Air flow patterns
8. Environmental monitoring & Personnel monitoring program
9. Equipment design
10. Container closure system
11. Media fill process (Aseptic process simulation) etc.
Most observations are related to smoke studies, media fills and aseptic behavior of operating people. While the first two are addressed by scientifically designing the process and qualification program, the behavior aspect requires intense training. Apart from detailed training program using audio visual techniques the training program should involve actual working in a pilot aseptic facility mimicking actual facility. The qualification process must be rigorous and should include –
1. Gowning qualification
2. Observing behavior of the operator under different scenarios like assembly, cleaning, disinfection, interventions, handling of abnormalities etc.
3. Personnel monitoring after the activity on multiple days
4. Participating in media fill trial (actually performing the activity the person will be doing in live scenario)
Other areas which require strengthening
Quality of documents
Apart from the five areas discussed earlier, companies need to significantly improve the quality of documents like standard operating procedures, master batch records, validation protocols, validation reports, APQRs. Responsibilities for creation, review and approval of these important documents have been delegated at lower levels in the organization without assessing capabilities of the people. This has resulted in drop in the quality of these documents. Often, these documents make little sense to the reader and auditors are forced to ask multiple questions to understand what is written. With remote inspections becoming the norm, this is going to pose serious risk. Levels for review and approval need to be standardized and people signing the documents must be held accountable for gaps identified in internal audits and Gemba walks. Gemba walks and internal audits should focus on verifying the activities being performed and matching them with the written documents. All SOPs should be qualified before implementing by actually observing each step while being executed and matching with the written procedure.
Process validation
Although the new guideline on process validation became effective in 2011, some companies are continuing old practice of executing three batches to complete validation without focusing on adequacy of process design and developing process understanding. On paper they demonstrate completion of three stages of the validation as required by the revised guideline but very few really understand the true intent of the guideline in achieving the goal of each of the step. For instance,
1. At the design stage the process needs to be fully established leaving nothing to the judgement of the operators
2. At process qualification stage
a. Suitability of equipment for intended process need to be established and
b. In PPQ batches, ability of process to consistently produce desired outcome needs to be demonstrated through extensive sampling (heightened level of scrutiny). The data need to be statistically evaluated to demonstrate uniformity within the batch and in between the batches.
c. The higher level of scrutiny needs to be continued beyond PPQ batches till enough confidence is gained about the process.
3. Continued process verification is not another document to be created at periodic interval but it is continual review (trend analysis) of CMAs, CPPs & CQAs to ensure that the process remains under control. Any out of trend values need to be investigated to understand the impact on Quality and identify changes required of any in the control strategy.
This concept of Design, Qualification and continued verification applies to not just manufacturing process but every process like water purification process, cleaning process, sterilization processes, etc.
Automation, Digitization, Data analytics, AI, IoT, PAT
It is very important to adopt new technologies for continuous improvement. Automation & digitization certainly helps in improving process consistency by eliminating human errors and gathering real time data related to the process and activities. Lot of focus needs to be given while designing automated or digitized processes.
Data analytics, AI, IoT etc. should be used to gain more understanding about the process. These tools should be used to support the core scientific knowledge and not as replacement. When these tools are used for running Quality relevant processes or taking Quality decisions, it should be ensured that they are fully validated. While many times deployment of such tools is done by external experts or consultants, people at the site should be able to understand the design fully and should be able to explain the same to the auditors.
People capability
Many of my earlier recommendations would remain as wishful thinking unless there are adequate number of qualified people with thorough knowledge of pharmaceutical technology apart from other relevant skills. Pharmaceutical companies need to deeply engage with pharmacy colleges to improve the quality of pharmacy education.
Conclusion
Concerted efforts are required from every company to improve the compliance level. Companies need to move away from mere compliance to guideline to Quality improvement. This requires developing in-depth understanding of everything we do and move away from superficiality.


Excellent compilation with the actual inderstanding of the working in a pharma company.
ReplyDeleteThe article explains in detail what ails pharma industries that results in deviations from set procedure and standards. Some of these problems are due lack of capabilities in the process developer who couldn't foresee the limitations of the facility or operator. Solutions for problems identified are well listed and can help industry to move towards perfection.
ReplyDeleteThis compilation contains alll the major aspects where the WLs are high.
ReplyDeleteThe control over these listed points will reduce the observations and keeping the indian industry on top of compliance
Thank you Mr. Ganadhish Kamat for an incisive and comprehensive analysis of deficiencies leading to weak compliance. There is a take away for all hierarchial levels of organisation.
ReplyDeleteMy elaboration on some of your points is as below :-
A. Production officers and their superiors, many a time, confine themselves to delivering production numbers of volume and time. Consistent quality and true compliance is not a priority for many of them. So there is no thinking in that direction from them.
Their understanding of compliance is limited to documentation completion.
B. QA officers and superiors limit themselves to procedural compliance and clean, correct documentation alone without a thought to understanding design intent of a procedure.
This sometimes results in an irrational or exaggerated fear of what an auditor would think.
Both above result in superficiality as summed up at the end of article.
B. Investigations teams many a time treat investigations as a formality to be completed so that operation is not interrupted. This results in weak superficial investigations.
The cardinal principle -" every coincidence is not necessarily a co-relation and every co-relation is not necessarily a cause" is forgotten when investigating. A first-seen-coincidence is clutched at like a straw and conclusions drawn.
D. Good learning opportunities reside in "Rejections" analysis on a regular basis and study of "Out-of-trend" occurrences whether on positive side or negative. But if the approach is to give precedence to "urgent but not so important" over "important but not urgent " then such opportunities are wasted. Deeper knowledge of process does not come about.
Lastly, the point to ponder over is why India who once was ahead of China in regulatory understanding has now fallen behind China in score ? This requires analysis by higher level hierarchy and it may provide insight to corrective measures at a broader level.
Comment made by: Vilas Satpute
DeleteMy association with Mr. Kamat has been over 15 years. His articles and talks are always on a practical approach with execution to near perfection and this article is yet another utilitarian to the industry.
ReplyDeleteThis article reveals an astonishing certitude that Indian site quality score was 6.8 which is below RoW and China!
While the Indian pharmaceutical industry made commendable progress on the quality front over the last decade, it's a well begun journey towards excellence but time to accelerate the speed.
Key takeaways
A. Shop-floor team both production and quality should focus more on value added themes and remove NVAs - Gemba walks by seniors would help in inducing on-the-job training and knowledge sharing to the younger folks
B. Serious and thoughtful efforts on root cause establishment - Need of the hour
C. QbD should become a cornerstone for R&D folks while developing the products- No look-back once the product is validated
D. Continue efforts on automation and digitization- Less or zero manual intervention.
Sir
ReplyDeleteExcellent and comprehensive compilation by considering current trends in the industry.
As usual very useful information from you again.
Thanks for it
Facts nicely put together, if any organization puts more focus on these areas, more than 80% audit readiness is taken care.
ReplyDeletepharmaceutical regulatory consulting is an indispensable resource for the industry, providing strategic guidance and actionable insights to enhance overall compliance, quality, and global competitiveness.
ReplyDeleteVery comprehensive information and need deeply thinking on current pharmaceutical companies approach.
ReplyDeleteVery good article with relevancy of day to day practical approach. Things in pharmaceutical industries are very simple but there is missing deep dive approach that's creat mismatch.
ReplyDeleteTHANKS ,REALL ITS COMPREHENSIVE INSIGHTS
ReplyDelete