Why Quality of medicines in India can not be taken for granted

- Ganadhish Kamat
Introduction
India is considered as the Pharmacy of the world and supplies high quality medicines to countries world over. While Indian drug manufacturers are capable of supplying high quality medicines at low price, can all the medicines available in India be considered to be of same quality? First of all, how do you define the quality of medicines?
Applying simple definition of Quality, "Fit for the purpose", a good quality medicine should produce desired effect after administration (Efficacy) without producing any undesired & harmful effect (Safety). Incase of products such as apparels, footwear, consumer products, food articles, electronics etc, consumers can judge the quality themselves based on sensory evaluation or evaluation of performance by actual use. If they are not happy with the quality, they have choice of not buying the product again or even sometimes returning it. This however is not possible for consumers of medicines (patients). Except in case of few medicines where there are visible symptoms, patient won't know whether the medicine is effective or not unless diagnostic tests are performed. In some cases they may get the effect but that may not be optimal. In case of emergency use medicines, there is no second chance and if the medicine doesn't have desired effect, the patient may loose the life or develop disability. It is even more difficult to detect undesired effects. For example carcinogenic or genotoxic impurities if present in the drug may show their effect long after the treatment is stopped. Presence of microbial contamination in injections may cause serious health hazard but such contamination comes to light when it is too late. Because of such high severity of impact of poor quality of medicines, it is imperative to establish adequate controls on the quality of medicines.
Quality of medicines
As mentioned earlier Efficacy and Safety are two facets of Quality of medicines.
Efficacy of the medicines depend upon many aspects such as presence correct drug (active ingredient) in right amount as stated on the label, rate of dissolution or drug release after administration, rate of absorption of the drug in the blood stream to maintain right amount of plasma concentration etc. The rate of dissolution or release and the rate of absorption depends on the inactive ingredients (excipients) used in the formulation, their grades, particle sizes, quantities, sequence of addition, uniform distribution, various processing parameters etc. Sometime the drug may exhibit polymorphism (present in two or multiple crystal structures with varying orientations or conformations). In may cases only certain form may be active and other may be inactive or even toxic. For example L-propranolol is an effective beta blocker, while D-propranolol is not. Incase of anti tuberculosis drug Ethambutol, the (S,S)-(+)-enantiomer is effective, while the (R,R)-(-)-enantiomer can cause blindness. So the presence of right amount of drug in the tablet or capsule is not enough to demonstrate efficacy of the drug. The efficacy of the dosage form (tablet/capsule/syrup/injection/eye drop etc) requires -
- Right drug in right form
- Correct amount as per the label
- Right combination of excipients (correct quantities, grades, particle sizes etc)
- Correct processing as per the validated process (correct order of addition, uniform distribution, process parameters as per the pre-established validated limits)
- Right packaging
- Proper Storage and Transportation conditions
- Absence or rare occurrence of serious adverse reactions and low incidence of other undesired actions from the drug itself
- Absence of unwanted form of the drug or presence below safety level established by toxicity studies.
- Control of any impurities below limits established by tox studies, or levels specified in ICH guideline.
- Process impurities - Impurities generated during the manufacturing of the drug such as -
- Unreacted starting materials
- Metallic impurities coming from the equipment, starting materials and catalysts used in the process
- By-products
- Drug excipient adducts
- Residual solvents
- External contamination / cross contamination - Such impurities may include -
- Particulate & other contamination from equipment, environment, personnel, processing aids, raw materials etc
- Cross-contamination with traces of other drug made using same area/equipment due to inadequate cleaning of area and equipment or faulty design of area and air handling system.
- Microbial contamination from environment, personnel etc due to poor facility or process design, inadequate gowning, inadequate cleaning etc.
- Degradation products - All drugs are prone for degradation on storage or handling due to exposure to heat, moisture, light, oxidative agents, acidic or alkaline environment. Such degradation results in fall in purity (potency) of the drug and increase in impurity levels. Such impurities are called degradation products.
- The drug present in the generic drug product is identical (same chemical structure, same form) to that in the innovator medicine.
- The drug product does not contain impurities higher than those found in innovator medicines or levels prescribed in ICH standards throughout its shelf life (demonstrated by conducting stability study under controlled conditions). If any new impurity is identified due to difference in the process, its safety levels are established by performing toxicity studies in animals.
- In case of orally administered drugs the product is bioequivalent (plasma concentration achieved with respect to C-Max and AUC are similar) to the innovator drug.
- In case of products such as injections, eye/ear drops etc equivalence is demonstrated by comparing the formulations with the innovator.
- Description
- Identification
- Solubility
- Optical rotation
- Polymorph
- Water content
- Assay (purity/potency)
- Organic Impurities
- Chiral impurities
- Residual solvents
- Metallic impurities
- Particle size distribution
- Microbial limit tests
- Endotoxin (If intended for use in injections)
- Description
- Identification
- Dissolution rate
- Water content
- Assay
- Impurities (Degradation products)
- Residual solvents (If solvents are used in the process)
- Microbial limit tests
Drug approval process & Regulatory controls on quality in advanced countries (and most of the developing countries)
- Complete characterization of the drug substance,
- Impurity profiles,
- Scientifically sound specifications for the drug substance and drug product,
- Validated test methods which have been demonstrated to be stability indicating,
- Specifications and source of the excipients used,
- Detailed formulation (composition) and manufacturing procedures including critical control parameters for the drug substance and the drug product,
- Data of forced degradation studies,
- Multi-media dissolution profiles ,
- Summary of drug development (Critical Quality attributes, Critical material attributes, Critical Process parameters, Control strategy etc),
- Exhibit (test) batch records including results of all test conducted on the same,
- Stability data for the drug substance and drug product generated as per the climatic zone and at accelerated conditions,
- Data & controls to demonstrate sterility assurance incase of sterile products,
- Data of Clinical trials / Bio-equivalence studies,
- Packaging configuration and packaging material specifications
Drug approval process & Regulatory controls on quality in India
Unlike most of the countries, Indian regulatory system follows dual approach. As per the Indian drug laws, there are two drug regulatory organizations, one at the center called Central Drug Standards Control Organization (CDSCO) and other at the states, Food and Drug Administration (FDA). These Central and State agencies fall under respective health ministries and hence are largely independent of each other. Their responsibilities also differ quite a bit. Under the Drug and Cosmetics Act, the CDSCO is responsible for approval of New Drugs, Clinical Trials in the country, laying down the standards for Drugs, control over the quality of imported Drugs, coordination of the activities of State Drug Control Organizations and providing expert advice with a view to bring about uniformity in the enforcement of the Drugs and Cosmetics Act. CDSCO is also responsible for approval of licenses of specified categories of Drugs such as blood and blood products, I. V. Fluids, Vaccine and Sera. On the other hand the state FDA is responsible for regulation of manufacture, sale and distribution of Drugs in their state. In addition state FDA has dual role of controlling the quality of food. Thus while CDSCO is responsible for new drug approval, state FDA is responsible for granting license for manufacture of drugs which are more than 4 year old in the country. This 4 year clause is quite irrational and unscientific.
For the new drug approval, CDSCO is required to follow the process described in NDCT rules, which is largely aligned with the process followed by advanced countries. The quality of the review however is questionable looking at the some of the approvals granted for new drugs in recent past, type of deficiencies identified in the review process. There is also no rigorous pre-approval inspection to verify the authenticity of the data submitted in the application. This is evident from the fact that while foreign regulatory agencies such as USFDA identifies significant violations in Indian firms including, data falsification, back dating, destruction of GMP documents, compromises in sterility assurance etc, inspections conducted by Indian regulators rarely identify them. There is also lack of transparency as the results of clinical trials and rationale for approval etc are not available for public scrutiny.
The process followed by the State FDAs is worse. First of all there are no written rules which they are required to follow for grant of license. This results in wide variability in the process followed in different states. To illustrate this I have given below a table to compare the documents a manufacturer is required to submit for obtaining manufacturing license in different states.
Documents required | AP | TS | MH | Gujrat | MP | HP | J&K | Sikkim |
Cover Letter | Y | Y | N | Y | N | Y | Y | Y |
Form 24/27 | Y | Y | N | Y | Y | Y | Y | Y |
Challan/Fees | Y | Y | Y | Y | Y | Y | Y | Y |
Copy of approval status from DCG(I)/IP 2010/M&M permission | Y | Y | Y | Y | Y | Y | Y | Y |
Copy of valid manufacturing license | Y | Y | N | N | N | Y | Y | Y |
Copy of valid GMP certificate | Y | Y | N | N | N | * | N | N |
Composition (unit dose) | Y | Y | Y | N | N | * | N | N |
Brief manufacturing process and flow chart | Y | Y | N | Y | Y | * | N | N |
Specifications and method of analysis of raw materials | Y | Y | N | N | N | * | N | N |
Specifications and method of analysis of Finished product | Y | Y | Y | Y | Y | * | N | N |
List of equipment (Both QC and Manufacturing) | Y | Y | N | N | N | * | N | N |
Draft specimen label | Y | Y | Y | Y | N | * | Y | Y |
List of technical staff | Y | Y | N | N | N | * | N | N |
Consent letters of technical staff and copies of their approvals (1 QC and 1 manufacturing chemist) | Y | Y | N | N | N | * | N | N |
Stability Data with valid T license copy | Y | Y | Y | N | Y | * | Y | N |
Copy of authorization letter | Y | Y | N | Y | N | Y | N | N |
Production block Details | Y | N | N | N | N | N | N | N |
* To be provided only if requested.
Apart from the lack of uniformity in the requirements for documents to be submitted to seek product permission, the list of documents indicate that there is very little technical evaluation done at the time of granting the manufacturing license. For instance, most important documents which are required to be reviewed to assure quality, efficacy & safety of the product, namely the product development report, analytical method validation reports, justification of specification, BA/BE study report etc are not part of requirement in any of the states. Although 9th amendment to D&C rules dated 3rd April 2017, made it mandatory to submit BE results for BCS class II & IV drugs, same has not been implemented in most states. For the products which are not official in Pharmacopoeia, the manufacturer can get away by setting their own specifications which may not be in line with ICH requirements. It is not mandatory to include important tests such as dissolution rate for solid oral dosage forms and test for related substances, metallic impurities and residual solvents. Since it is allowed to have shelf life limit of not less than 90% of label claim for the drug content, the product may show degradation as high as 10% and still meet the specification. The degradation products formed may produce severe adverse effects if not controlled under established safety limits. In absence of BA/BE study showing that the drug product is bioequivalent to Innovator product or Reference Listed Drug there is no guarantee that the product will produce same effect as seen in the clinical trials. If the drug product or the drug substance is official in Indian Pharmacopoeia or other recognized Pharmacopoeia, the drug law mandates compliance to the official monographs. This however is not adequate to assure quality. The Pharmacopeial monographs are minimum standards a drug is required to comply with. They however don't take into account the manufacturing processes of different manufacturers which can generate different impurities as mentioned earlier. So to assure quality it is important establish suitability of Pharmacopeial monograph by carefully evaluating the manufacturing process. It may be noted that the drugs manufactured in one state can be freely sold in entire country so the FDA in a state has no control over the quality of medicines being sold in their states.
Control of changes
Storage and distribution
Recalls
Good Manufacturing practices & Regulatory inspections
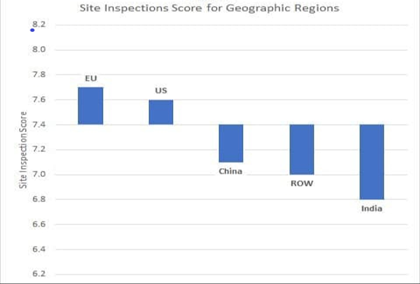
While scientific evaluation of development data and control measures by regulators at the time of granting approval is essential for ensuring that quality is built into design, adherence to Good manufacturing practices (GMPs) is essential to make sure that the manufacturing and related activities are carried out as per the design and in adherence to scientifically established systems and procedures so as to ensure consistent good quality of medicines. Adherence to good manufacturing activities also helps in preventing contamination of drugs from external contaminants, cross-contamination from other drugs manufactured in the facility, mix-ups etc. For example, if the manufacturer of cough syrups mentioned earlier had complied to GMPs, contamination with DEG would not have occurred thereby saving lives of so many innocent children. Adherence to GMPs is verified by regulatory agencies by conducting periodic inspections of the manufacturing sites. Regulatory agencies of advanced countries like USA, UK, EU, TGA etc mostly conduct such inspections once in two years. The exact frequency & duration of inspection is decided based on risk assessment and past inspection history. The duration of inspections vary from 5-15 days with 1-3 inspectors depending upon the complexity of the site.Indian law requires inspections of manufacturing sites to be conducted by state FDA once in a year. Such inspections are carried out by 1-2 inspectors over 1-2 days. Obviously these inspections are quite inadequate to detect serious GMP deficiencies. Couple of years back CDSCO started risk based inspections of manufacturing sites. As per last report, CDSCO conducted inspection of around 400 manufacturing sites. Approximately 1/3rd of these sites were found to be operating with serious violations of GMPs warranting their shutdown. This clearly shows inadequacy of the inspections by state FDA. India has around 10,000 manufacturing sites most of these are run by MSMEs. Some of these are manufacturing drugs for large scale companies under contract manufacturing agreement. If we extrapolate findings of CDSCO inspections to all sites, we can expect more than 3000 sites to be operating in violation to GMP norms. At the pace of 400 inspections per year, it will take forever for CDSCO to inspect all the facilities. India has around 600 manufacturing sites approved by USFDA. Most of these are operated by large scale companies and are expected to have better compliance as compared to other sites. USFDA inspections have many times identified serious GMP violations including falsification of data, destruction of GMP documents, risk of cross-contamination, inadequate controls on sterility assurance, inadequate investigations etc. resulting in actions such issue of warning letter or import alert. I don't remember seeing any similar actions taken by Indian regulators (central or state) on these sites. Unlike USFDA where inspection reports are available to public through Freedom of information (FOI), Indian regulators do not share inspection reports. So it is difficult to assess the depth of inspections, kind of deficiencies identified and kind of risks they pose to public health.
Conclusion

While many pharmaceutical firms in India have high ethical and quality standards and establish scientifically sound controls to assure quality of medicines, some of the manufacturers do bare minimum to merely comply with the laws of the land while some even take shortcuts in adhering to already lax laws. Indian citizens can not be left to the mercy of the manufacturers to provide high quality drugs. India's drug regulatory system needs to be made robust by way of making the drug laws in line with global standards and their strict implementation to ensure that every drug sold in the country meets same high standard of safety and quality.

Quite comprehensive and so well written. Thank you Ganadhish for your efforts to bring in required change.
ReplyDeleteVery well explained in details. Hope Regulators and Politicians take note and start acting appropriately.
ReplyDeleteHorrible... Not very true
ReplyDeleteIs it a thesis without proper data... Ridiculous
ReplyDeleteVery well written and comprehensive article on pharma quality. Can be used as an introductory chapter of a text book. And certainly useful for giving orientation to young managers. Brings out FDA working anomalies qyite clearly.
ReplyDeleteSir elaborate article first time saw some one caring about medicine supply for Indian public
ReplyDeleteVery nicely articulated artical ever seen on pharma quality. Government should use such ppl who served many years for industry and has zeal to work for quality.
ReplyDeleteWell written and comprehensive article
ReplyDeleteVery detailed blog and covers all the aspects of quality so well .
ReplyDeleteHats-off to you for bringing out the drug regulatory flaws in India so well. Indian citizens surely deserve better quality medicines, and it’s high time that our policymakers pay attention to these.
ReplyDeletevery informative sir.....Indian Drug Authority has to still travel miles of distance to reach to standard of US or UK drug authorities.
ReplyDeleteVery insightful,still a long way to go India is rated high for medicine (are safe!)supplier to globe despite of absence of our presence in PICs or other MR health authorities
ReplyDelete